
Galactosemia
Deficiência de galactose-1-fosfato uridil transferase; Deficiência de galactoquinase; Deficiência de galactose-6-fosfato epimeraseA galactosemia é um distúrbio no qual o corpo não consegue transformar (metabolizar) galactose em glicose.
Causas
A galactosemia é uma doença hereditária, isto é, passada de geração em geração. Se cada um dos pais carrega uma cópia não funcionante do gene que causa a galactosemia, cada filho tem uma chance de 25% (1 em 4) de ser afetado pela doença.
Existem três formas da doença:
- Deficiência da enzima galactose-1-fosfato uridil transferase (galactosemia clássica, a forma mais comum e mais grave)
- Deficiência de galactoquinase
- Deficiência de galactose-6-fosfato epimerase
As pessoas com galactosemia não conseguem transformar o açúcar simples da galactose. A galactose compõe metade da lactose, o açúcar encontrado no leite.
Se um bebê com galactosemia tomar leite, substâncias feitas de galactose se acumularão em seu sistema. Essas substâncias danificam o fígado, cérebro, rins e olhos.
Pessoas com galactosemia não toleram qualquer tipo de leite (humano ou animal). Elas devem ter cuidado com a ingestão de outros alimentos que contenham galactose.
Sintomas
Bebês com galactosemia podem desenvolver sintomas nos primeiros dias de vida se consumirem leite materno ou qualquer outro alimento que contenha lactose. Os sintomas podem ser devido a uma infecção grave no sangue com a bactéria E. coli.
Os sintomas incluem:
-
Convulsões
Convulsões
Crises convulsivas são achados físicos ou alterações no comportamento que ocorrem após um episódio de atividade elétrica anormal no cérebro. O termo ...
Ler artigo agora Adicionar a favoritos -
Irritabilidade
Irritabilidade
Crianças pequenas que ainda não sabem falar comunicam quando algo está errado agindo de forma confusa ou irritável. Se o seu filho está mais irritad...
 ImagenLer artigo agora Adicionar a favoritos
ImagenLer artigo agora Adicionar a favoritos -
Letargia
Letargia
Fadiga é uma sensação de desgaste, cansaço ou falta de energia.
Ler artigo agora Adicionar a favoritos - Dificuldade de alimentação (o bebê se recusa a tomar mamadeira contendo leite)
- Ganho de peso insuficiente
- Pele e olhos amarelados (icterícia)
- Vômitos
Sinais e exames
Os exames incluem:
- Hemocultura para infecção bacteriana (sepse por E. coli)
- Atividade enzimática nos glóbulos vermelhos
- Cetonas na urina
- Diagnóstico pré-natal ao medir diretamente a enzima galactose-1-fosfato uridil transferase
- Substâncias redutoras na urina do bebê e nível normal ou baixo de açúcar no sangue, enquanto o recém-nascido está sendo alimentado com leite materno ou com leite em pó contendo lactose
Baixo de açúcar no sangue
Hipoglicemia é uma condição que ocorre quando o nível de açúcar (glicose) no sangue encontra-se muito baixo. Quando o nível de açúcar no sangue é men...
 ImagenLer artigo agora Adicionar a favoritos
ImagenLer artigo agora Adicionar a favoritos
Exames de triagem neonatal podem incluir triagem para diagnóstico precoce da galactosemia.
Resultados dos exames podem mostrar:
- Aminoácidos na urina ou no sangue
-
Aumento do fígado
Aumento do fígado
Hepatomegalia é o inchaço do fígado além do seu tamanho normal. Se tanto o fígado como o baço estiverem aumentados, isso é chamado de hepatoesplenome...
 ImagenLer artigo agora Adicionar a favoritos
ImagenLer artigo agora Adicionar a favoritos -
Fluido abdominal
Fluido abdominal
Ascite é o acúmulo de líquido no espaço entre os tecidos que revestem o abdome e os órgãos abdominais.
 ImagenLer artigo agora Adicionar a favoritos
ImagenLer artigo agora Adicionar a favoritos -
Hipoglicemia
Hipoglicemia
Hipoglicemia é uma condição que ocorre quando o nível de açúcar (glicose) no sangue encontra-se muito baixo. Quando o nível de açúcar no sangue é men...
ImagenLer artigo agora Adicionar a favoritos
Tratamento
As pessoas com galactosemia devem evitar todo tipo de leite e produtos que contenham leite (inclusive leite em pó), além de qualquer alimento que contenha galactose, por toda a vida. É essencial ler os rótulos dos produtos e ser um consumidor bem informado.
Os bebês podem ser alimentados com:
- Leite de soja
- Leites a base de carne ou fórmula de hidrolisado de proteína
- Outro leite sem lactose
Suplementos de cálcio são recomendados.
Expectativas (prognóstico)
Pessoas com diagnóstico precoce e que evitam produtos lácteos totalmente podem viver uma vida relativamente normal. No entanto, uma leve incapacidade intelectual pode se desenvolver, mesmo em pessoas que evitam galactose.
Complicações
Complicações incluem:
-
Catarata
Catarata
A catarata é uma opacidade do cristalino, a lente natural do olho.
 ImagenLer artigo agora Adicionar a favoritos
ImagenLer artigo agora Adicionar a favoritos -
Cirrose do fígado
Cirrose
A cirrose caracteriza-se pela fibrose (formação de tecido fibroso em substituição às células hepáticas) e consequente funcionamento deficiente do fíg...
 ImagenLer artigo agora Adicionar a favoritos
ImagenLer artigo agora Adicionar a favoritos - Atraso no desenvolvimento da fala
- Períodos menstruais irregulares, diminuição da função dos ovários levando à falência ovariana
- Retardo mental
- Infecção bacteriana grave (sepse por E. coli)
- Tremores e funções motoras incontroláveis
- Morte (se houver galactose na dieta)
Quando contatar um profissional de saúde
Ligue para o médico se:
- Seu bebê tiver sintomas de galactosemia
- Se você tiver histórico familiar de galactosemia e estiver considerando ter filhos
Prevenção
Conhecer seu histórico familiar ajuda muito. Se tiver um histórico familiar de galactosemia e desejar ter filhos, o aconselhamento genético ajudará a tomar decisões sobre gravidez e exames pré-natais. Uma vez diagnosticada a galactosemia, o aconselhamento genético é recomendado para outros membros da família.
Alguns exames de triagem neonatal podem verificar se há galactosemia. Se o teste indicar um possível diagnóstico de galactosemia, é recomendado eliminar produtos lácteos da dieta do bebê imediatamente e entrar em contato com o médico para realização de um exame de sangue confirmatório.
Referências
Berry GT. Classic galactosemia and clinical variant galactosemia. 2000 Feb 4 [Updated 2017 Mar 9]. In: Pagon RA, Adam MP, Ardinger HH, et al, eds. GeneReviews [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2017. PMID: 20301691. www.ncbi.nlm.nih.gov/books/NBK1518/.
Bonnardeaux A, Bichet DG. Inherited disorders of the renal tubule. In: Skorecki K, Chertow GM, Marsden PA, Taal MW, Yu ASL, eds. Brenner and Rector's The Kidney. 10th ed. Philadelphia, PA: Elsevier; 2016:chap 45.
Broomfield A, Brain C, Grunewald S. Galactosaemia: diagnosis, management and long-term outcome. Paediatrics and Child Health. 2015:25(3);113-118.
Gibson KM, Pearl PL. Inborn errors of metabolism and the nervous system. In: Daroff RB, Jankovic J, Mazziotta JC, Pomeroy SL. Bradley's Neurology in Clinical Practice. 7th ed. Philadelphia, PA: Elsevier; 2016:chap 91.
Kishnani PS, Chen Y-T. Defects in metabolism of carbohydrates. In: Kliegman RM, Stanton BF, St. Geme JW, Schor NF, eds. Nelson Textbook of Pediatrics. 20th ed. Philadelphia, PA: Elsevier; 2016:chap 87.
Maitra A. Diseases of infancy and childhood. Kumar V, Abbas AK, Aster JC, eds. Robbins and Cotran Pathologic Basis of Disease. 9th ed. Philadelphia, PA: Elsevier Saunders; 2015:chap 10.
-
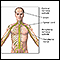
Galactosemia - Ilustração
Um bebê com galactosemia não é capaz de usar (metabolizar) o açúcar simples da galactose, que alcança níveis altos no corpo, causando lesões no fígado, no sistema nervoso central e em vários outros sistemas do organismo. Um bebê com galactosemia pode desenvolver icterícia, vômitos, letargia, irritabilidade e convulsões.
Galactosemia
Ilustração

-
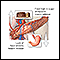
Galactosemia - Ilustração
Um bebê com galactosemia não é capaz de usar (metabolizar) o açúcar simples da galactose, que alcança níveis altos no corpo, causando lesões no fígado, no sistema nervoso central e em vários outros sistemas do organismo. Um bebê com galactosemia pode desenvolver icterícia, vômitos, letargia, irritabilidade e convulsões.
Galactosemia
Ilustração
Data da revisão: 4/24/2023
Revisão feita por: Anna C. Edens Hurst, MD, MS, Associate Professor in Medical Genetics, The University of Alabama at Birmingham, Birmingham, AL. Review provided by VeriMed Healthcare Network. Also reviewed by David C. Dugdale, MD, Medical Director, Brenda Conaway, Editorial Director, and the A.D.A.M. Editorial team.
© 1997- A.D.A.M., uma unidade de negócios da Ebix, Inc. Qualquer duplicação ou distribuição das informações aqui contidas é estritamente proibida.
 Todos os direitos reservados
Todos os direitos reservados