
Síndrome de Marfan
Aneurisma de aorta - MarfanA síndrome de Marfan é uma doença do tecido conjuntivo, o tecido que fortalece as estruturas do corpo.
As doenças do tecido conjuntivo afetam o sistema esquelético, o sistema cardiovascular, os olhos e a pele.
Causas
A síndrome de Marfan é causada por defeitos em um gene chamado fibrilina-1, que desempenha um papel importante como componente do tecido conjuntivo no corpo.
O defeito genético também causa crescimento excessivo dos ossos longos do corpo. Pessoas com esta síndrome têm uma altura elevada, com os braços e pernas longos. Não se sabe bem como esse supercrescimento acontece.
Outras áreas do corpo que são afetadas incluem:
- Tecido pulmonar (pode haver um pneumotórax, no qual o ar poderá escapar do pulmão para a cavidade torácica, provocando colapso do pulmão)
- A aorta, o principal vaso sanguíneo que leva o sangue do coração para o corpo, pode estar anormalmente elástica ou ter a parede enfraquecida (dilatação ou aneurisma de aorta)
- Os olhos, o que pode causar catarata e outros problemas (como o descolamento do cristalino)
- A pele
- O tecido que cobre a medula espinhal
Na maioria dos casos, a síndrome de Marfan é herdada, o que significa que ela passada de geração para geração. No entanto, até 30% dos pacientes não têm histórico familiar. Esses casos são chamados de "esporádicos". Nestes casos, acredita-se que a síndrome seja causada por um novo defeito genético espontâneo.
Sintomas
As pessoas com a síndrome de Marfan costumam ser altas com pernas e braços longos e finos, e dedos como o de uma aranha (aracnodactilia). Quando esticam os braços, a envergadura é maior do que a altura.
Outros sintomas incluem:
- Tórax afundado ou saliente, chamado tórax em funil (pectus excavatum) ou peito de pombo (pectus carinatum)
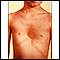
Pectus excavatum
Peito escavado descreve uma formação anormal da caixa torácica que dá ao tórax uma aparência cavada ou afundada.
 ImagenLer artigo agora Adicionar a favoritos
ImagenLer artigo agora Adicionar a favoritosPectus carinatum
Peito carinado descreve uma protrusão do tórax sobre o esterno, cujas descrições frequentemente mencionam uma pessoa com aparência de ave.
 ImagenLer artigo agora Adicionar a favoritos
ImagenLer artigo agora Adicionar a favoritos - Pés chatos
- Palato muito arqueado ou apinhamento dental
-
Hipotonia
Hipotonia
Hipotonia significa a diminuição do tônus muscular.
 ImagenLer artigo agora Adicionar a favoritos
ImagenLer artigo agora Adicionar a favoritos - Articulações muito flexíveis (mas os cotovelos poderão ser menos flexíveis)
- Problemas de aprendizado
- Movimento do cristalino do olho em relação à sua posição normal (deslocamento)
- Miopia
- Mandíbula inferior pequena (micrognatia)
- Coluna vertebral curvada para um lado (escoliose)
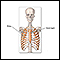
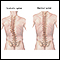
Escoliose
A escoliose é um encurvamento da coluna vertebral. A coluna vertebral é a série de ossos no meio das costas. Há normalmente alguma curvatura na col...
 ImagenLer artigo agora Adicionar a favoritos
ImagenLer artigo agora Adicionar a favoritos - Rosto fino e estreito
Muitas pessoas com síndrome de Marfan têm dor crônica nos músculos e nas articulações.
Sinais e exames
O médico realizará um exame físico. Poderão ser observados articulações hipermóveis e sinais de:
-
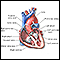
Aneurisma
Aneurisma
Um aneurisma é um alargamento irregular de uma porção de uma artéria causado por fraqueza nas paredes do vaso sanguíneo. Um aneurisma da aorta toráci...
 ImagenLer artigo agora Adicionar a favoritos
ImagenLer artigo agora Adicionar a favoritos - Pneumotórax
- Problemas em valvas cardíacas
Um exame ocular poderá mostrar:
- Defeitos na lente ou na córnea
-
Deslocamento da retina
Deslocamento da retina
O descolamento de retina é a separação da membrana fotossensível na parte de trás do olho (a retina) das suas camadas de suporte.
 ImagenLer artigo agora Adicionar a favoritos
ImagenLer artigo agora Adicionar a favoritos - Problemas de visão
Os seguintes testes podem ser realizados:
- Ecocardiograma
- Teste da mutação da fibrilina-1 (em algumas pessoas)
Um ecocardiograma deve ser feito uma vez por ano para analisar a base da aorta e possíveis problemas cardíacos valvares.
Tratamento
Quando possível, os problemas de visão devem ser tratados.
Monitore a escoliose, principalmente durante a adolescência.
Medicamentos para diminuir os batimentos cardíacos podem diminuir o estresse na aorta. Evite participar de competições esportivas e praticar esportes de contato para não ter problemas cardíacos. Algumas pessoas podem precisar de cirurgia para substituir a base e a valva aórtica.
As pessoas com síndrome de Marfan devem tomar antibióticos antes de procedimentos odontológicos para evitar endocardite (infecção das valvas). Mulheres grávidas com a síndrome de Marfan devem ser monitoradas com atenção devido ao aumento do estresse no coração e na aorta.
Expectativas (prognóstico)
Complicações cardíacas podem diminuir a expectativa de vida de pessoas com essa doença. No entanto, muitos pacientes vivem bem até aos 60 anos ou mais. Tratamento adequado e cirurgia podem prolongar ainda mais a expectativa de vida.
Complicações
As complicações podem incluir:
- Regurgitação aórtica
- Ruptura aórtica
- Endocardite bacteriana
- Aneurisma dissecante da aorta
- Alargamento da base da aorta
-
Insuficiência cardíaca
Insuficiência cardíaca
A insuficiência cardíaca, também chamada de insuficiência cardíaca congestiva, é uma doença na qual o coração não consegue mais bombear sangue sufici...
ImagenLer artigo agora Adicionar a favoritos - Prolapso da válvula mitral
- Escoliose
- Problemas de visão
Quando contatar um profissional de saúde
Casais que têm essa condição e que estão planejando ter filhos podem querer realizar um aconselhamento genético antes de começar uma família.
Prevenção
Novas mutações genéticas espontâneas causando síndrome de Marfan (menos de 1/3 dos casos) não podem ser evitadas. Se você tiver a síndrome de Marfan, consulte com seu médico pelo menos uma vez por ano.
Referências
Doyle A, Doyle JJ, Dietz HC. Marfan syndrome. In: Kliegman RM, Stanton BF, St Geme JW, Schor NF, eds. Nelson Textbook of Pediatrics. 20th ed. Philadelphia, PA: Elsevier; 2016:chap 702.
Pyeritz RE. Inherited diseases of connective tissue. In: Goldman L, Schafer AI, eds. Goldman's Cecil Medicine. 24th ed. Philadelphia, PA: Elsevier Saunders; 2016:chap 260.
-
Pectus excavatum - Ilustração
Tórax escavado é uma condição na qual o "osso do peito" (esterno) aparenta estar afundado e o tórax côncavo. Às vezes, é chamada de "tórax afunilado". A maioria dos casos não está associada a nenhuma outra condição (casos isolados). Entretanto, algumas síndromes podem provocar o tórax escavado.
Pectus excavatum
Ilustração
-
Síndrome de Marfan - Ilustração
A síndrome de Marfan é um distúrbio do tecido conjuntivo que causa anomalias esqueléticas tipicamente reconhecidas em uma pessoa muito alta e magra. A pessoa com a síndrome de Marfan pode exibir membros longos e dedos em formato de aranha, anomalias do tórax, curvatura da coluna e um conjunto específico de características faciais que incluem palato muito arqueado e dentes apinhados. Os defeitos mais significativos da síndrome são as anomalias cardiovasculares, que podem incluir aumento (dilatação) da base da aorta. Como a síndrome de Marfan geralmente é uma doença hereditária, futuros pais com um histórico familiar de síndrome de Marfan devem receber aconselhamento genético.
Síndrome de Marfan
Ilustração


-
Pectus excavatum - Ilustração
Tórax escavado é uma condição na qual o "osso do peito" (esterno) aparenta estar afundado e o tórax côncavo. Às vezes, é chamada de "tórax afunilado". A maioria dos casos não está associada a nenhuma outra condição (casos isolados). Entretanto, algumas síndromes podem provocar o tórax escavado.
Pectus excavatum
Ilustração
-
Síndrome de Marfan - Ilustração
A síndrome de Marfan é um distúrbio do tecido conjuntivo que causa anomalias esqueléticas tipicamente reconhecidas em uma pessoa muito alta e magra. A pessoa com a síndrome de Marfan pode exibir membros longos e dedos em formato de aranha, anomalias do tórax, curvatura da coluna e um conjunto específico de características faciais que incluem palato muito arqueado e dentes apinhados. Os defeitos mais significativos da síndrome são as anomalias cardiovasculares, que podem incluir aumento (dilatação) da base da aorta. Como a síndrome de Marfan geralmente é uma doença hereditária, futuros pais com um histórico familiar de síndrome de Marfan devem receber aconselhamento genético.
Síndrome de Marfan
Ilustração
Data da revisão: 5/5/2016
Revisão feita por: Michael A. Chen, MD, PhD, Associate Professor of Medicine, Division of Cardiology, Harborview Medical Center, University of Washington Medical School, Seattle, WA. Also reviewed by David Zieve, MD, MHA, Isla Ogilvie, PhD, and the A.D.A.M. Editorial team.
© 1997- A.D.A.M., uma unidade de negócios da Ebix, Inc. Qualquer duplicação ou distribuição das informações aqui contidas é estritamente proibida.
 Todos os direitos reservados
Todos os direitos reservados