
Talassemia
Anemia mediterrânea; Anemia de Cooley; Betatalassemia; AlfatalassemiaA talassemia é uma doença sanguínea herdada entre os membros de uma família (hereditária) na qual o organismo produz uma forma anormal de hemoglobina. A hemoglobina é a proteína encontrada nos glóbulos vermelhos que carrega o oxigênio. Essa doença resulta em destruição excessiva dos glóbulos vermelhos, causando anemia.
Causas
A hemoglobina é formada por duas cadeias de proteínas:
- Alfa
- Beta
A talassemia ocorre quando há um defeito em um gene que ajuda a controlar a produção de uma dessas proteínas.
Existem dois tipos principais de talassemia:
- A alfatalassemia ocorre quando um gene ou genes relacionados à cadeia alfa está ausente ou sofreu uma alteração (mutação).
- A betatalassemia ocorre quando defeitos genéticos similares afetam a produção da cadeia beta.
As alfatalassemias ocorrem mais comumente em pessoas originárias do sudoeste asiático, do Oriente Médio, da China e afrodescendentes.
As betatalassemias ocorrem em indivíduos originários da região do mediterrâneo e, em menor extensão, chineses, demais asiáticos e afrodescendentes.
Existem muitas formas de talassemia. Cada tipo possui muitos subtipos diferentes. Tanto a alfatalassemia quanto a betatalassemia incluem as duas formas abaixo:
- Talassemia maior
- Talassemia menor
É necessário herdar o gene defeituoso de ambos os pais para desenvolver a talassemia maior.
A talassemia menor ocorre se o indivíduo receber o gene defeituoso somente de um dos pais. Indivíduos com essa forma da doença são portadores do distúrbio, mas geralmente não apresentam sintomas.
A betatalassemia maior também é denominada anemia de Cooley.
Os fatores de risco para a talassemia incluem:
- Etnia asiática, chinesa, mediterrânea ou africana
- Histórico familiar da doença
Sintomas
A forma mais grave de alfatalassemia maior causa natimorto (morte do feto durante o parto ou nos últimos estágios da gravidez).
Crianças que nascem com betatalassemia maior (anemia de Cooley) são normais até o nascimento, mas desenvolvem anemia grave durante o primeiro ano de vida.
Anemia
A anemia é uma doença em que há uma quantidade insuficiente de glóbulos vermelhos (hemácias) saudáveis. Os glóbulos vermelhos fornecem oxigênio para...
Outros sintomas podem incluir:
- Deformidades ósseas na face
-
Fadiga
Fadiga
Fadiga é uma sensação de desgaste, cansaço ou falta de energia.
Ler artigo agora Adicionar a favoritos - Crescimento retardado
-
Falta de ar
Falta de ar
Dificuldade para respirar pode envolver:Dificuldade respiratóriaRespiração desconfortávelSensação de não receber ar em quantidade suficiente
 ImagenLer artigo agora Adicionar a favoritos
ImagenLer artigo agora Adicionar a favoritos - Pele amarela (icterícia)
Indivíduos com a forma menor de alfa e betatalassemia apresentam glóbulos vermelhos pequenos, mas nenhum sintoma.
Sinais e exames
Um exame físico pode revelar aumento do baço.
Uma amostra de sangue é coletada e enviada a um laboratório para ser examinada.
- Os glóbulos vermelhos parecerão pequenos e com formato anormal quando observados ao microscópio.
- Um hemograma completo revela anemia.
- Um exame denominado eletroforese de hemoglobina identifica a presença de uma forma anormal de hemoglobina.
- Um exame denominado análise mutacional pode ajudar a detectar a alfatalassemia.
Tratamento
O tratamento para talassemia maior geralmente envolve transfusões sanguíneas regulares e suplementos de folato.
Caso receba transfusões de sangue, o indivíduo não deve tomar suplementos de ferro. Isso poderia acarretar um aumento grande da quantidade de ferro no organismo, o que pode ser prejudicial.
Indivíduos que recebem um número significativo de transfusões sanguíneas necessitam de um tratamento denominado terapia de quelação, para eliminar o excesso de ferro no organismo.
Um transplante de medula óssea pode ajudar a tratar a doença em alguns pacientes, especialmente em crianças.
Expectativas (prognóstico)
A talassemia grave pode causar morte prematura devido à insuficiência cardíaca, geralmente os entre 20 e 30 anos de idade. Transfusões sanguíneas regulares e terapia para eliminar o ferro do organismo ajudam a melhorar o prognóstico.
Formas menos graves de talassemia, em geral, não diminuem a expectativa de vida.
Aconselhamento genético e exames de pré-natal podem ajudar indivíduos com histórico familiar dessa condição que estejam planejando ter filhos.
Complicações
A talassemia maior não tratada causa insuficiência cardíaca e problemas no fígado, além de deixar o indivíduo mais suscetível a desenvolver infecção.
Transfusões sanguíneas podem ajudar a controlar alguns sintomas. Entretanto, elas podem resultar em uma quantidade excessiva de ferro.
Quando contatar um profissional de saúde
Entre em contato com o seu médico se:
- Você ou seu filho apresentar sintomas de talassemia
- Estiver recebendo tratamento para a doença e novos sintomas se desenvolverem
Referências
Cappellini MD. The thalassemias. In: Goldman L, Schafer AI, eds. Goldman's Cecil Medicine. 25th ed. Philadelphia, PA: Elsevier Saunders; 2016:chap 162.
DeBaun MR, Frei-Jones MJ, Vichinsky EP. Hemoglobinopatiies. In: Kliegman RM, Stanton BF, St Geme JW, Schor NF, eds. Nelson Textbook of Pediatrics. 20th ed. Philadelphia, PA: Elsevier; 2016:chap 462.
Giardina PJ, Rivella S. Thalassemia syndromes. In: Hoffman R, Benz EJ Jr, Silberstein LE, Heslop HE, Weitz JI, Anastasi J, eds. Hematology: Basic Principles and Practice. 6th ed. Philadelphia, PA: Elsevier Saunders; 2013:chap 38.
-
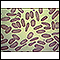
Talassemia maior - Ilustração
Talassemia major é uma forma hereditária de anemia hemolítica caracterizada pelas anormalidades de produção das hemácias (hemoglobinas). Esta é a forma mais grave de anemia e a depleção de oxigênio no corpo se torna aparente nos primeiros 6 meses de vida. Se não tratada, geralmente há ocorrência de óbito em alguns anos. Observe as hemácias pequenas, pálidas (hipocrômicas) e com formato anormal associadas à talassemia major. As células mais escuras provavelmente representam as hemácias normais de uma transfusão de sangue.
Talassemia maior
Ilustração
-
Talassemia menor - Ilustração
Talassemia minor é uma forma hereditária de anemia hemolítica que é menos severa que a talassemia major. Este teste no sangue de um indivíduo com talassemia apresenta hemácias pequenas (microcítica), pálidas (hipocrômica) e com formatos variados (poiquilocitose). As hemácias pequenas conseguem levar menos oxigênio que as hemácias normais.
Talassemia menor
Ilustração


-
Talassemia maior - Ilustração
Talassemia major é uma forma hereditária de anemia hemolítica caracterizada pelas anormalidades de produção das hemácias (hemoglobinas). Esta é a forma mais grave de anemia e a depleção de oxigênio no corpo se torna aparente nos primeiros 6 meses de vida. Se não tratada, geralmente há ocorrência de óbito em alguns anos. Observe as hemácias pequenas, pálidas (hipocrômicas) e com formato anormal associadas à talassemia major. As células mais escuras provavelmente representam as hemácias normais de uma transfusão de sangue.
Talassemia maior
Ilustração
-
Talassemia menor - Ilustração
Talassemia minor é uma forma hereditária de anemia hemolítica que é menos severa que a talassemia major. Este teste no sangue de um indivíduo com talassemia apresenta hemácias pequenas (microcítica), pálidas (hipocrômica) e com formatos variados (poiquilocitose). As hemácias pequenas conseguem levar menos oxigênio que as hemácias normais.
Talassemia menor
Ilustração
Data da revisão: 2/12/2016
Revisão feita por: Todd Gersten, MD, Hematology/Oncology, Florida Cancer Specialists & Research Institute, Wellington, FL. Review provided by VeriMed Healthcare Network. Also reviewed by David Zieve, MD, MHA, Isla Ogilvie, PhD, and the A.D.A.M. Editorial team.
© 1997- A.D.A.M., uma unidade de negócios da Ebix, Inc. Qualquer duplicação ou distribuição das informações aqui contidas é estritamente proibida.
 Todos os direitos reservados
Todos os direitos reservados