Mucopolisacaridosis tipo I

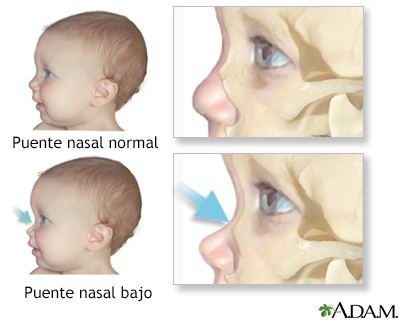
Puente nasal bajo
Programme Una Cita
Definición
La mucopolisacaridosis tipo I (MPS I) es una rara enfermedad en la cual el cuerpo carece o no tiene suficiente cantidad de una enzima necesaria para descomponer cadenas largas de moléculas de azúcar. Estas cadenas son llamadas glucosaminoglucanos (anteriormente denominados mucopolisacáridos). Como resultado, las moléculas se acumulan en diferentes partes del cuerpo y causan diversos problemas de salud.
Esta afección pertenece a un grupo de enfermedades llamado mucopolisacaridosis (MPS). MPS I es el tipo más común
Existen varios tipos de MPS, incluyendo:
Causas
La MPS I es hereditaria, lo cual significa que sus padres normalmente le transmiten la enfermedad a usted. Si ambos padres portan una copia inactiva del gen relacionado con esta afección, cada uno de sus hijos tiene un 25% (1 en 4) de probabilidades de desarrollar la enfermedad.
Las personas con MPS I no producen una enzima llamada alfa-L-iduronidasa lisosómica. Esta enzima ayuda a descomponer las cadenas largas de moléculas de azúcar llamadas glucosaminoglucanos. Estas moléculas se encuentran en todo el cuerpo, a menudo en las secreciones mucosas y en el líquido que rodea las articulaciones.
Sin la enzima, los glucosaminoglucanos se acumulan y causan daño a órganos, incluso al corazón. Los síntomas pueden ir de leves a graves. La forma leve se denomina MPS I atenuada y la forma severa se denomina MPS I grave.
Síntomas
Los síntomas de MPS I casi siempre aparecen de los 3 a los 8 años de edad. Los niños con MPS I grave presentan síntomas antes que aquellos con la forma menos grave.
Algunos de los síntomas incluyen:
- Huesos anormales en la columna
- Incapacidad de abrir completamente la mano (mano en garra)
- Córneas opacas
- Sordera
- Crecimiento interrumpido
- Problemas de válvula cardíaca
- Enfermedad articular, incluso rigidez
- Discapacidad intelectual que empeora con el tiempo en la MPS I grave
- Rasgos faciales gruesos y toscos con puente nasal bajo
Pruebas y exámenes
En algunos estados, a los bebés se les examina en busca de MPS I como parte de las pruebas de detección para recién nacidos.
Otros exámenes que se pueden hacer dependiendo de los síntomas incluyen:
- Electrocardiograma ECG
- Pruebas genéticas para buscar cambios en el gen de la alfa-L-iduronidasa (IDUA)
- Exámenes de orina en busca de mucopolisacáridos adicionales
- Radiografía de la columna
Tratamiento
La terapia de reemplazo enzimático se puede recomendar. El medicamento llamado laronidasa (Aldurazyme), se administra a través de una vena (IV, vía intravenosa). Reemplaza la carencia de la enzima. Hable con el proveedor de atención médica de su hijo para obtener más información.
Se ha utilizado un trasplante de médula ósea. El tratamiento ha tenido resultados mixtos.
Otros tratamientos dependen de los órganos que están afectados.
Grupos de apoyo
Se puede encontrar más información y apoyo para las personas con MPS I y sus familias en:
- National MPS Society -- www.mpssociety.org
- National Organization for Rare Disorders -- rarediseases.org/rare-diseases/mucopolysaccharidosis-type-i
- NIH Genetic and Rare Disorders Information Center -- rarediseases.info.nih.gov/diseases/10335/mucopolysaccharidosis-type-i
Expectativas (pronóstico)
Los niños con MPS I grave por lo general no tienen un buen pronóstico. Sus problemas de salud empeoran con el tiempo, resultando en la muerte a la edad de 10 años.
Los niños con MPS I atenuada (más leve) tienen menos problemas de salud, muchos con vidas bastante normales que llegan a la adultez.
Cuándo contactar a un profesional médico
Comuníquese con su proveedor si:
- Tiene antecedentes familiares de MPS I y está considerando la posibilidad de tener hijos
- Su hijo empieza a mostrar síntomas de MPS I
Prevención
Los expertos recomiendan la asesoría genética y la realización de exámenes para parejas con antecedentes familiares de MPS I que estén contemplando la posibilidad de tener hijos. Hay disponibilidad de pruebas prenatales.
Referencias
Kumar V, Abbas AK, Aster JC. Genetic disorders. In: Kumar V, Abbas AK, Aster JC, eds. Robbins & Cotran Pathologic Basis of Disease. 10th ed. Philadelphia, PA: Elsevier; 2021:chap 5.
Lampe C. Mucopolysaccharidoses. In: Kliegman RM, St. Geme JW, Blum NJ, et al, eds. Nelson Textbook of Pediatrics. 22nd ed. Philadelphia, PA: Elsevier; 2025:chap 109.
Pyeritz RE. Inherited diseases of connective tissue. In: Goldman L, Cooney KA, eds. Goldman-Cecil Medicine. 27th ed. Philadelphia, PA: Elsevier; 2024:chap 239.
Turnpenny PD, Ellard S, Cleaver R. Inborn errors of metabolism. In: Turnpenny PD, Ellard S, Cleaver R, eds. Emery's Elements of Medical Genetics and Genomics. 16th ed. Philadelphia, PA: Elsevier; 2022:chap 18.

Actualizado : 4/8/2025
Versión en inglés revisada por : Anna C. Edens Hurst, MD, MS, Associate Professor in Medical Genetics, The University of Alabama at Birmingham, Birmingham, AL. Review provided by VeriMed Healthcare Network. Also reviewed by David C. Dugdale, MD, Medical Director, Brenda Conaway, Editorial Director, and the A.D.A.M. Editorial team.
La información aquí proporcionada no debe utilizarse durante ninguna emergencia médica ni para el diagnóstico o tratamiento de ninguna afección médica. Se debe consultar a un profesional médico autorizado para el diagnóstico y tratamiento de cualquiera y todas las afecciones médicas. Los enlaces a otros sitios se proporcionan únicamente con fines informativos; no constituyen una recomendación de dichos sitios. No se ofrece garantía alguna, expresa ni implícita, en cuanto a la precisión, fiabilidad, puntualidad o exactitud de las traducciones realizadas por un servicio externo de la información aquí proporcionada a cualquier otro idioma.
© 1997-
A.D.A.M., una unidad de negocio de Ebix, Inc. Queda estrictamente prohibida la duplicación o distribución de la información aquí contenida.
Todo el contenido de este sitio, incluyendo texto, imágenes, gráficos, audio, video, datos, metadatos y compilaciones, está protegido por derechos de autor y otras leyes de propiedad intelectual. Usted puede ver el contenido para uso personal y no comercial. Cualquier otro uso requiere el consentimiento previo por escrito de Ebix. Usted no puede copiar, reproducir, distribuir, transmitir, mostrar, publicar, realizar ingeniería inversa, adaptar, modificar, almacenar más allá del almacenamiento en caché habitual del navegador, indexar, hacer minería de datos, extraer o crear obras derivadas de este contenido. Usted no puede utilizar herramientas automatizadas para acceder o extraer contenido, incluyendo la creación de incrustaciones, vectores, conjuntos de datos o índices para sistemas de recuperación. Se prohíbe el uso de cualquier contenido para entrenar, ajustar, calibrar, probar, evaluar o mejorar sistemas de inteligencia artificial (IA) de cualquier tipo sin el consentimiento expreso por escrito. Esto incluye modelos de lenguaje grandes, modelos de aprendizaje automático, redes neuronales, sistemas generativos, sistemas de recuperación aumentada y cualquier software que ingiera contenido para producir resultados. Cualquier uso no autorizado del contenido, incluyendo el uso relacionado con la IA, constituye una violación de nuestros derechos y puede dar lugar a acciones legales, daños y sanciones legales en la medida en que lo permita la ley. Ebix se reserva el derecho de hacer valer sus derechos mediante medidas legales, tecnológicas y contractuales.