Síndrome de Apert

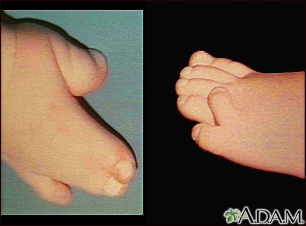
Sindactilia
Programme Una Cita
Definición
Es una enfermedad genética en la cual las suturas entre los huesos del cráneo se cierran más temprano de lo normal. Esto afecta la forma de la cabeza y la cara. Los niños con el síndrome de Apert a menudo también tienen deformidades en las manos y en los pies.
Causas
El síndrome de Apert se puede trasmitir de padres a hijos (hereditario), como un rasgo autosómico dominante. Esto significa que solo uno de los padres necesita transmitir el gen alterado para que un hijo tenga la afección.
La mayoría de los casos se pueden presentar sin un antecedente familiar conocido.
El síndrome de Apert es causado por uno de dos cambios en el gen FGFR2. Este cambio en el gen provoca que algunas de las suturas óseas del cráneo se cierren demasiado temprano. Esta afección se conoce como craneosinostosis.
Síntomas
Los síntomas incluyen:
- Cierre prematuro de las suturas entre los huesos del cráneo, que se nota por la formación de crestas a lo largo de las suturas (craneosinostosis)
- Infecciones frecuentes del oído
- Fusión o unión marcada del segundo, tercero y cuarto dedos de las manos, que regularmente se conoce como "manos en mitón"
- Pérdida de la audición
- Punto blando grande o de cierre tardío en el cráneo del bebé (fontanela)
- Desarrollo intelectual posiblemente lento (varía de una persona a otra)
- Ojos prominentes o abultados
- Subdesarrollo grave de la parte media de la cara
- Anomalías esqueléticas (de las extremidades)
- Estatura baja
- Fusión o unión de los dedos de los pies
Algunos otros síndromes que incluyen craneosinostosis pueden llevar a una apariencia similar de la cara y la cabeza, pero no incluyen los problemas graves de manos y pies del síndrome de Apert. Estos síndromes similares incluyen:
- Síndrome de Carpenter (Kleeblattschädel, deformidad del cráneo en forma de cruce en trébol)
- Enfermedad de Crouzon (disostosis craneofacial)
- Síndrome de Pfeiffer
- Síndrome de Saethre-Chotzen
Pruebas y exámenes
Su proveedor de atención médica llevará a cabo un examen físico. Se tomarán radiografías del cráneo, la mano y el pie. Siempre se deben realizar audiometrías.
Las pruebas genéticas pueden confirmar el diagnóstico del síndrome de Apert.
Tratamiento
El tratamiento consiste en una cirugía para corregir el crecimiento óseo anormal del cráneo, al igual que para la fusión de los dedos de las manos y de los pies. Los niños con este trastorno deben ser examinados por un equipo especializado en cirugía craneofacial en un centro médico infantil.
Si hay problemas auditivos, se debe consultar con un especialista en este campo.
Grupos de apoyo
Puede encontrar más información y apoyo para personas con el síndrome de Apert y sus familias en:
- Children's Craniofacial Association: ccakids.org
Cuándo contactar a un profesional médico
Comuníquese con su proveedor si tiene antecedentes familiares del síndrome de Apert o si nota que el cráneo de su bebé no se está desarrollando normalmente.
Prevención
La asesoría genética puede ser valiosa si usted tiene un antecedente familiar de este trastorno y está planeando quedar embarazada. Su proveedor puede evaluar a su bebé en busca de esta enfermedad durante el embarazo.
Referencias
Goldstein JA, Davit AJ, Losee JE. Pediatric plastic surgery. In: Zitelli BJ, McIntire SC, Nowalk AJ, Garrison J, eds. Zitelli and Davis' Atlas of Pediatric Physical Diagnosis. 8th ed. Philadelphia, PA: Elsevier; 2023:chap 23.
Kinsman SL, Johnston MV. Congenital anomalies of the central nervous system. In: Kliegman RM, St. Geme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM, eds. Nelson Textbook of Pediatrics. 21st ed. Philadelphia, PA: Elsevier; 2020:chap 609.
Mauck BM. Congenital anomalies of the hand. In: Azar FM, Beaty JH, eds. Campbell's Operative Orthopaedics. 14th ed. Philadelphia, PA: Elsevier; 2021:chap 80.
National Center for Advancing Translational Sciences. Genetic and Rare Diseases Information Center website. Acrocephalosyndactyly type I. rarediseases.info.nih.gov/diseases/5833/apert-syndrome. Updated September 2025. Accessed October 15, 2025.

Actualizado : 9/18/2023
Versión en inglés revisada por : Anna C. Edens Hurst, MD, MS, Associate Professor in Medical Genetics, The University of Alabama at Birmingham, Birmingham, AL. Review provided by VeriMed Healthcare Network. Also reviewed by David C. Dugdale, MD, Medical Director, Brenda Conaway, Editorial Director, and the A.D.A.M. Editorial team.
La información aquí proporcionada no debe utilizarse durante ninguna emergencia médica ni para el diagnóstico o tratamiento de ninguna afección médica. Se debe consultar a un profesional médico autorizado para el diagnóstico y tratamiento de cualquiera y todas las afecciones médicas. Los enlaces a otros sitios se proporcionan únicamente con fines informativos; no constituyen una recomendación de dichos sitios. No se ofrece garantía alguna, expresa ni implícita, en cuanto a la precisión, fiabilidad, puntualidad o exactitud de las traducciones realizadas por un servicio externo de la información aquí proporcionada a cualquier otro idioma.
© 1997-
A.D.A.M., una unidad de negocio de Ebix, Inc. Queda estrictamente prohibida la duplicación o distribución de la información aquí contenida.
Todo el contenido de este sitio, incluyendo texto, imágenes, gráficos, audio, video, datos, metadatos y compilaciones, está protegido por derechos de autor y otras leyes de propiedad intelectual. Usted puede ver el contenido para uso personal y no comercial. Cualquier otro uso requiere el consentimiento previo por escrito de Ebix. Usted no puede copiar, reproducir, distribuir, transmitir, mostrar, publicar, realizar ingeniería inversa, adaptar, modificar, almacenar más allá del almacenamiento en caché habitual del navegador, indexar, hacer minería de datos, extraer o crear obras derivadas de este contenido. Usted no puede utilizar herramientas automatizadas para acceder o extraer contenido, incluyendo la creación de incrustaciones, vectores, conjuntos de datos o índices para sistemas de recuperación. Se prohíbe el uso de cualquier contenido para entrenar, ajustar, calibrar, probar, evaluar o mejorar sistemas de inteligencia artificial (IA) de cualquier tipo sin el consentimiento expreso por escrito. Esto incluye modelos de lenguaje grandes, modelos de aprendizaje automático, redes neuronales, sistemas generativos, sistemas de recuperación aumentada y cualquier software que ingiera contenido para producir resultados. Cualquier uso no autorizado del contenido, incluyendo el uso relacionado con la IA, constituye una violación de nuestros derechos y puede dar lugar a acciones legales, daños y sanciones legales en la medida en que lo permita la ley. Ebix se reserva el derecho de hacer valer sus derechos mediante medidas legales, tecnológicas y contractuales.